Smith-Waterman算法是一种用于生物信息学中序列比对的算法,特别是用于局部序列比对。它由Temple F. Smith和Michael S. Waterman在1981年提出。与全局比对算法(如Needleman-Wunsch算法)不同,Smith-Waterman算法专注于找出两个序列之间的最佳局部对齐,而不是整个序列的对齐。
一、基本概念
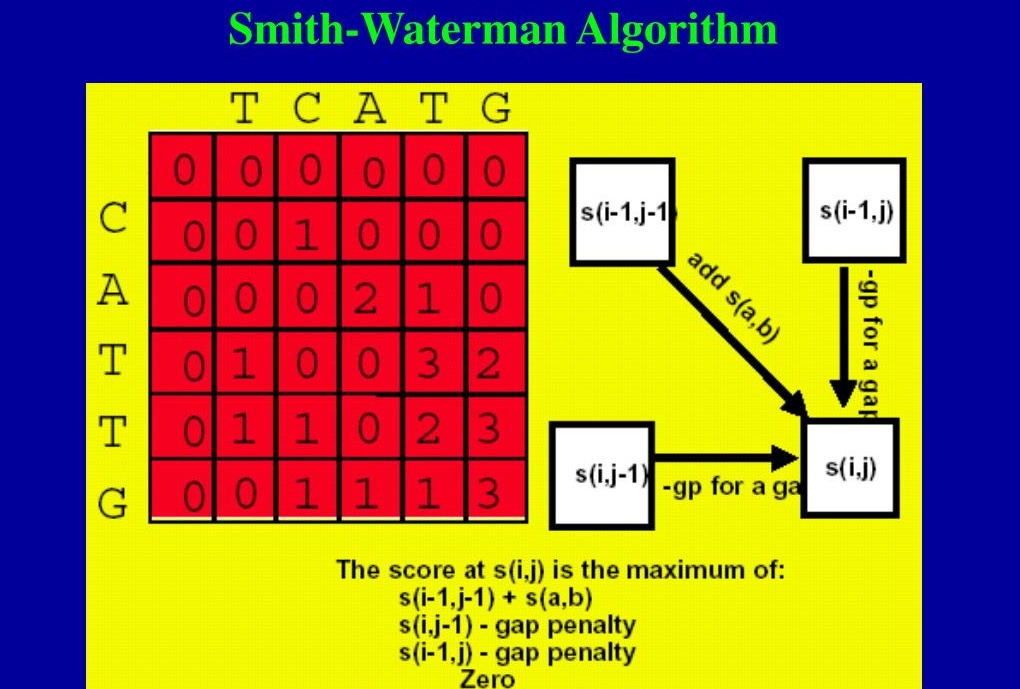
1. 分数矩阵(Scoring Matrix):算法首先创建一个分数矩阵,其大小为(m+1)×(n+1),m,n分别是两个序列的长度。矩阵的第一行和第一列通常初始化为0,表示没有序列参与比对的分数。
2. 匹配/不匹配分数:算法会根据匹配或不匹配的核苷酸或氨基酸给予不同的分数。通常,匹配的核苷酸或氨基酸会得到正分,而不匹配的会得到负分。
3. 间隙惩罚:当序列中的一个位置与另一个序列中的空位对齐时,算法会给予一定的负分,这被称为间隙惩罚。
4. 动态规划:Smith-Waterman算法使用动态规划来填充分数矩阵。通过比较当前位置的分数与左上角、上、左三个位置的分数,来决定当前位置的分数。
5. 局部对齐:算法的目标是找出得分最高的局部对齐,而不是整个序列的对齐。这意味着算法会追踪得分最高的路径,而不是填充整个矩阵。
6. 终止条件:算法在填充完分数矩阵后,会从得分最高的路径开始回溯,直到找到局部对齐的起始点。
7. 结果:最终,算法会输出两个序列之间得分最高的局部对齐区域,包括对齐的序列片段和对应的分数。
二、算法过程
Smith-Waterman算法的过程主要包括以下几个步骤:
1. 初始化得分矩阵:创建一个(m+1)×(n+1)矩阵,m,n分别分别是参与比对的两个序列的长度。矩阵的第一行和第一列初始化为0,表示序列开始的空位。
2. 定义匹配和不匹配分数:为匹配和不匹配的核苷酸或氨基酸分配分数。通常,匹配得正分,不匹配得负分。
3. 定义间隙罚分:为序列中引入间隙(插入或删除)的每个位置分配罚分。
4. 填充得分矩阵:使用动态规划算法,根据以下规则填充矩阵:
- 如果两个对应的元素相同或相似,则在矩阵的当前位置添加匹配分数。
- 如果不同,则可以选择添加不匹配分数或间隙罚分,取两者中的最大值。
- 如果当前位置的分数小于0,则将其设为0,表示该路径的比对结束。
5. 回溯找到局部最优解:从得分矩阵中的最大值开始回溯,直到矩阵中的值为0。回溯路径表示局部最优的序列对齐。
6. 输出结果:根据回溯路径,输出两个序列的局部对齐结果。
「」
回溯过程是从矩阵中的最大值开始,根据以下规则进行:
- 如果当前位置的值来自左上角,则说明当前两个序列的元素是匹配或不匹配的,记录下来,并移动到左上角的位置。
- 如果当前位置的值来自左侧或上方,则说明当前序列出现了间隙,记录下来,并移动到对应的位置。
- 如果当前位置的值是0,则回溯结束。
这个过程可以找到两个序列之间最相似的局部区域,而不是整个序列的全局对齐。
三、应用场景
Smith-Waterman算法主要应用于生物信息学领域,特别是在序列比对中寻找两个生物分子序列之间的局部相似性。以下是一些具体的应用场景:
1. 基因和蛋白质序列的局部比对:在研究基因或蛋白质的功能、结构和进化关系时,Smith-Waterman算法可以用来发现序列中高度保守或功能重要的局部区域。
2. 同源基因的识别:通过局部比对,可以识别不同生物体中具有相似功能的同源基因。
3. 蛋白质结构预测:在蛋白质结构预测中,通过局部比对可以发现已知结构的蛋白质与目标蛋白质之间的相似性,从而推测目标蛋白质可能的三维结构。
4. 基因组学研究:在基因组学研究中,Smith-Waterman算法可以用于识别基因组中的重复序列、转座子或其他基因组变异。
5. 疾病相关基因的发现:通过比对疾病相关序列与正常序列,Smith-Waterman算法有助于发现与疾病相关的基因变异。
6. 药物设计:在药物设计中,局部比对可以帮助识别与药物靶标相互作用的关键氨基酸残基,从而指导药物分子的设计。
7. RNA二级结构预测:Smith-Waterman算法也被用于RNA二级结构预测,通过比对已知的RNA结构来预测新的RNA分子的二级结构。
8. 算法优化和硬件加速:由于其在生物信息学中的重要性,Smith-Waterman算法的优化和硬件加速也是研究的热点,以提高大规模序列比对的效率。
这些应用场景展示了Smith-Waterman算法在现代生物学和医学研究中的广泛性和重要性。
四、优缺点
Smith-Waterman算法的优缺点如下:
### 优点:
1. 专注于局部最优比对:与全局比对算法不同,Smith-Waterman算法专注于寻找两个序列之间最相似的局部区域,这在很多生物学应用中非常有用。
2. 动态规划:算法使用动态规划技术,能够有效地找到序列之间的最佳局部对齐。
3. 灵活性:算法允许研究者自定义匹配、不匹配和间隙罚分,以适应不同的比对需求。
4. 适用性广:适用于蛋白质和核苷酸序列的比对,可以用于多种生物学问题,如基因表达分析、进化研究等。
5. 算法优化和硬件加速:存在多种优化版本的Smith-Waterman算法,以及针对特定硬件平台的加速实现,提高了算法的效率。
### 缺点:
1. 时间复杂度:尽管比全局比对算法快,但Smith-Waterman算法的时间复杂度仍然较高,特别是在处理较长序列时。
2. 空间复杂度:算法需要存储一个较大尺寸的分数矩阵,这可能导致在处理大规模数据时需要较多的内存资源。
3. 无法保证全局最优:由于算法的设计初衷是寻找局部最优解,因此它不能保证找到全局最优的比对结果。
4. 参数敏感性:算法的匹配分数、不匹配分数和间隙罚分对最终结果有很大影响,需要仔细选择这些参数以获得最佳比对结果。
5. 计算资源消耗:对于大规模数据集,算法可能需要大量的计算资源,这可能限制了其在资源受限的环境中的应用。
总的来说,Smith-Waterman算法是一种强大的工具,尤其适用于寻找序列间的局部相似性,但也需要考虑到其在计算时间和资源上的需求。
Smith-Waterman算法在生物信息学中非常有用,尤其是在寻找基因或蛋白质序列中的相似区域时。然而,由于它只关注局部对齐,因此在需要整个序列对齐的情况下,可能需要使用全局比对算法。